riboSeed
pipeline for using ribosomal flanking regions to improve bacterial genome assembly
![]()
riboSeed: Leveraging bacterial architecture to assemble across ribosomal regions
We have developed a genome assembly preprocessing scheme, riboSeed, that uses the unique regions flanking the ribosomal coding operons. Please give it a shot and let me know how it goes for you! If you love it, please tweet about it to #riboSeed; if you don’t like it, please send me a email instead :)
Preprint
the riboSeed preprint manuscript can be found on bioRxiv; comments are welcome!
Documentation
riboSeed’s full documentation can be found at http://riboseed.readthedocs.io.
Analysis scripts
The scripts used in the analyses in riboSeed’s preprint can be found on the gitHub page under scripts.
Validation Datasets
The script used to create the artificial genomes can be found here. The GAGE-B datasets can be download from their (website)[http://ccb.jhu.edu/gage_b/index.html]. The accessions for all other genomes used can be found in the supplemental information of the preprint.
Installation
riboSeed is available via the conda installation ecosystem using conda install riboseed (note the lowercase “s”). Alternatively, riboSeed can be installed via pip with pip install riboSeed. For required external tools, see the README.
Theory
rDNAs, the genomic regions containing the sequences coding for ribosomal RNAs, are often found multiple times in a single genome. Due to how well rDNA is conserved within a taxa, we hypothesized that if the regions flanking the rDNAs are sufficiently unique within a genome, those regions would be able to locate an rDNA within the genome during assembly.
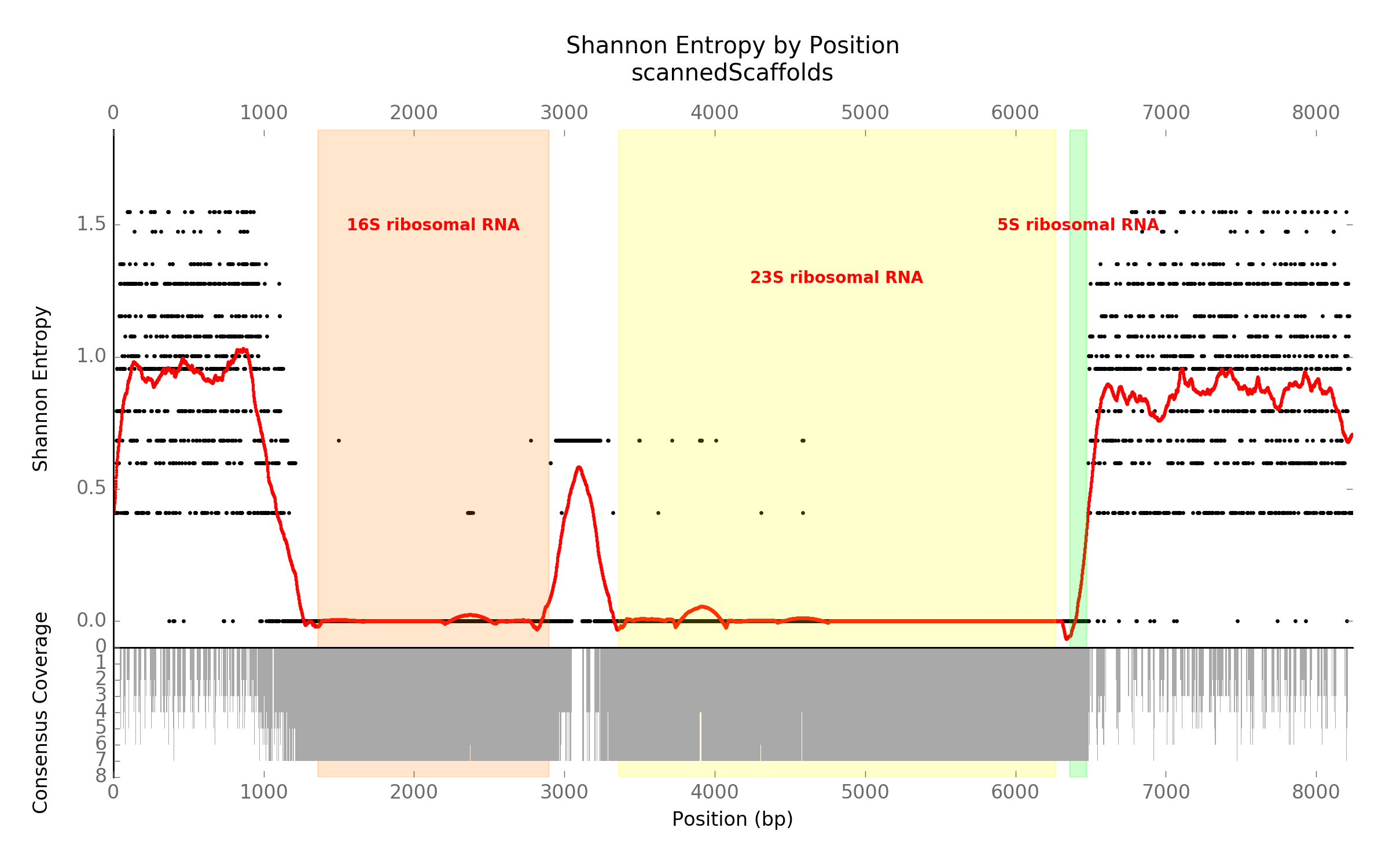 Here, we have extracted all 7 of the rDNA and 1kb flanking regions from the *E. coli Sakai * genome, aligned the sequences, and calculated the coverage and Shannon entropy for the alignments. This shows a high degree of conservation for the actual coding sequences, but sharply increasing entropy in the flanking regions immediately preceding and following. This shows that rDNAs, while having nearly identical coding sequences within a genome, have unique flanking regions.
Here, we have extracted all 7 of the rDNA and 1kb flanking regions from the *E. coli Sakai * genome, aligned the sequences, and calculated the coverage and Shannon entropy for the alignments. This shows a high degree of conservation for the actual coding sequences, but sharply increasing entropy in the flanking regions immediately preceding and following. This shows that rDNAs, while having nearly identical coding sequences within a genome, have unique flanking regions.
De Fere Novo Assembly
We call our method a de fere novo assembly, as we use a subassembly technique to minimize the bias caused by reference choice. We map the short reads to the reference genome, extract the reads mapping to rDNA (with flanking) regions, and perform subassemblies with SPAdes to reassemble the rDNA and flanking regions from the reads. These “long reads” are concatenated together separated with 5kb of N’s. The reads are then mapped to the concatenated sequence and and subassembled for several additional iterations.
Sample Dataset
We generated a simulated genome from the 7 rDNA regions with 5kb flanking regions, and then used ART (MountRainier-2016-06-05) to generated simulated MiSeq reads of various depths.
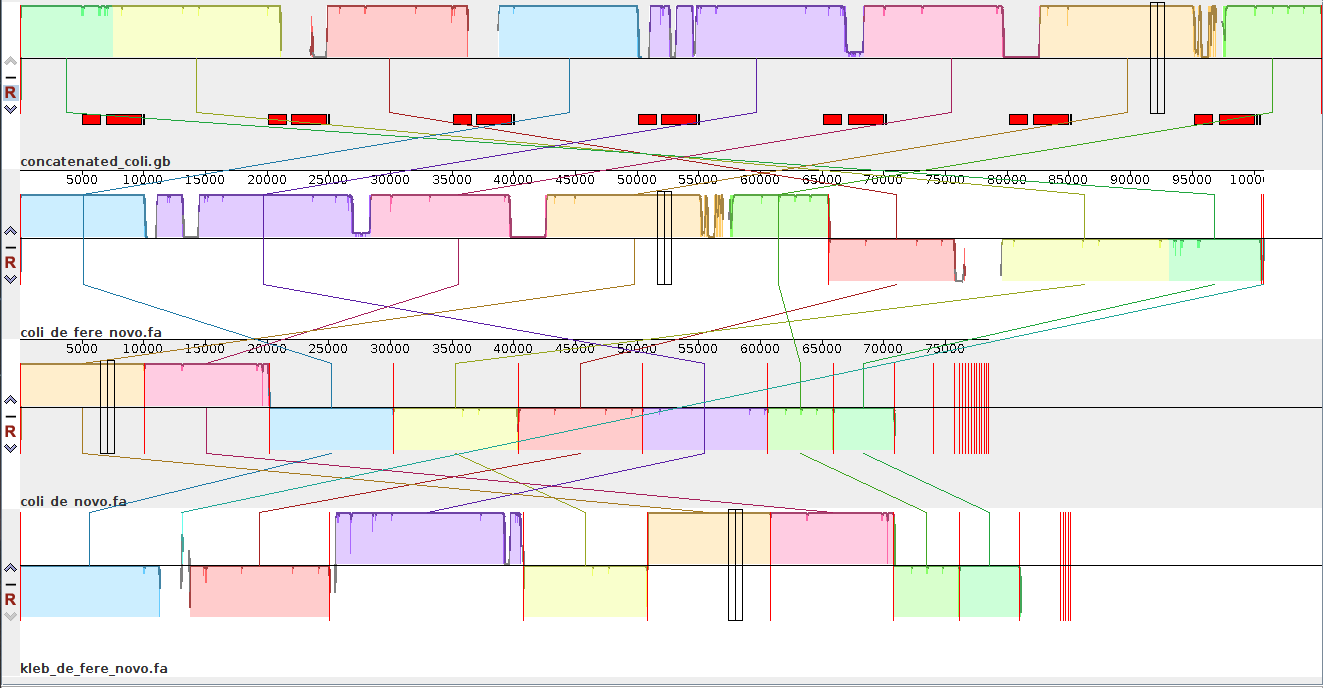
In this Mauve visualization, we show (from top to bottom) the reference simulated genome, riboSeed’s de fere novo assembly, de novo assembly, and a negative control de fere novo assembly using a Klebsiella reference genome. The results show that with riboSeed’s de fere novo assembly correctly joins six of the seven rDNA regions to reconstruct the simulated genome with only short reads. By contrast, the short reads alone failed to bridge any gaps caused by the repeated rDNAs, and the assembly using a poor reference choice only assembled across a single rDNA region. We have run this successfully on many real datasets with positive results
Conclusions
So what does this mean? We conclude that given short read sequencing data of sufficient (<10x) depth and a taxonomically close reference genome possessing sufficiently unique rDNA flanking regions, riboSeed’s de fere novo assembly can bridge across gaps in a de novo assembly caused by repeated rDNAs. Its not a silver bullet solving all short read assembly problems, but it reliably addresses a single issue affecting nearly all bacterial genome assemblies.
Further, when used in conjunction with the other genome finishing tools (namely, in a pipeline such as BugBuilder), riboSeed can result in closed genomes.
Next Steps
We are currently exploring applications to eukaryotic genome assembly, which can have tens or hundreds of rDNA.